7 Restart calculation
DS-PAW currently supports resuming calculations for structure relaxation, transition state calculations, molecular dynamics simulations, constant-potential calculations, and reading rho and wave functions. Users can specify file paths to read the final structure, magnetic moments, potential, and other relevant information from the previous calculation.
7.1 Continuation of Relax Calculation Instructions
In the event that the relaxation calculation is unexpectedly terminated, fails to converge within the maximum number of steps, or if a higher-precision relaxation calculation is desired, it is necessary to obtain the final structure from the previous calculation (including the magnetic moment information of the final configuration if spin is considered in the system) to perform the next relaxation calculation. In this case, the program will output latestStructure.as and relax.h5 files, both latestStructure.as and relax.h5 can be used as input files for a subsequent calculation. If you need to continue the calculation based on this structure, it is recommended to follow these steps:
Create a clean directory and prepare two input files: relax.in and latestStructure.as (or relax.h5);
Set the parameter sys.structure = latestStructure.as (or sys.structure = relax.h5) in the relax.in file. The name of the structure file can be modified, and it is recommended to provide a clear indication for the continuation calculation.
Submit the job for calculation.
latestStructure.as is one of the readable files for structure relaxation continuation calculations. In addition to this, the relax.h5 file can also be read as the final state structure.
7.2 NEB Transition State Calculation Continuation Instructions
If the transition state calculation is unexpectedly terminated, fails to converge within the maximum number of steps, or requires a higher accuracy calculation, you need to obtain the final structure from the previous calculation (including the magnetic moment information if the system considers spin) to perform the next transition state calculation. The transition state calculation involves multiple subfolders; in this case,
In each subfolder No, the files latestStructureNo.as and nebNo.h5 are output by default. The .as file can be used as the input file for a subsequent calculation. Taking an insertion point number of 3 as an example, if you need to continue the calculation based on this structure, you can directly call the NEB continuation script described in Auxiliary Tool User Guide:
Demonstration of the data processing procedure using a Python script:
Enter the directory of the initial NEB calculation and view the files in that directory:

Call the :guilabel:`neb_restart.py script in this directory and execute the following command:
1python neb_restart.py
Following the prompts in the interactive interface, specify the path to the original NEB file, the parameter file name, and the backup folder name. In this example, the backup folder is specified as bakfile.
Check the neb directory again:

Where bakfile is the backup file, and the 00-04 folders store the structure files required for resuming the calculation. You can submit directly within this directory to resume the calculation.
Backup folder bakfile structure analysis.
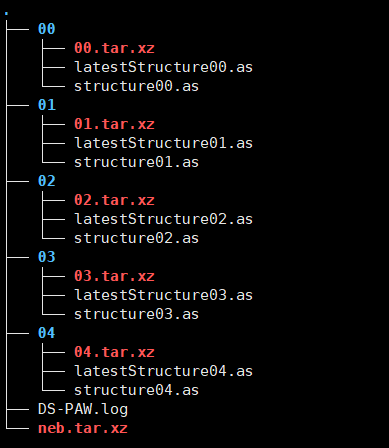
The outermost compressed archive, neb.tar.xz, in the backup folder contains the initial NEB calculation h5 files. The compressed archives in each subfolder contain backups of all files from the corresponding subfolder of the initial NEB calculation. The outermost layer outside the subfolders contains the initial and final state structure files from the initial calculation.
If users prepare input files themselves, it is recommended to follow the steps below:
Create a clean directory and place the neb.in file, initial and final structure files structure00.as and structure04.as, and the final structure files for intermediate configurations latestStructure01.as, latestStructure02.as, and latestStructure03.as into it;
Rename the intermediate structure files, :guilabel:`latestStructureNo.as, to :guilabel:`structureNo.as;
Create folders 00, 01, 02, 03, and 04, and place the corresponding structure files in each folder;
Submit the job for calculation.
The .as file is a readable file for continuing transition state calculations; it is not recommended to use the nebNo.h5 file as input for continuing calculations.
7.3 Instructions for Continuing AIMD Molecular Dynamics Simulations
If the molecular dynamics simulation was unexpectedly terminated, or if you wish to extend the simulation time, you need to obtain the final structure and velocities (and magnetization information for spin-polarized systems) from the previous calculation to perform a longer simulation. The molecular dynamics simulation by default outputs latestStructure.as and aimd.h5 file. Both latestStructure.as and aimd.h5 can be used as input files for continuation. If you need to continue the calculation from this structure, it is recommended to follow the steps below:
Create a clean directory and prepare two input files: aimd.in and latestStructure.as (or aimd.h5);
In the aimd.in file, set the parameter sys.structure = latestStructure.as (or sys.structure = aimd.h5). The name of the structure file can be modified; it is recommended to include a clear continuation run indication in the filename.
Submit the job for calculation.
latestStructure.as is one of the readable files for continuing molecular dynamics calculations, and besides that, the aimd.h5 file can also be read as the final structure.
Note:
To modify the ensemble for a continuation run, delete the information in the Next positions section of the latestStructure.as file; otherwise, the continuation run may result in errors.
7.4 Continuation Instructions for fixedPotential Calculations
The fixedPotential calculation uses the steepest descent method, which solves the target charge and potential values through multiple self-consistent iterations. This process can be viewed as n self-consistent calculations that depend on each other. If the calculation is unexpectedly interrupted before the charge converges, the continuation function can be used. This function uses the charge and potential values obtained before the interruption as the starting point to approach the target potential. The following steps are recommended to resume a constant potential calculation:
In the original calculation directory, modify the fixedPotential.in file to specify the directory containing the h5 file obtained from the initial calculation to resume the calculation. The corresponding parameter is
cal.iniFixedP = ./scf.h5.
Note:
If you want to keep the scf.h5 file from the initial calculation, you can rename the original file, such as renaming it to readscf.h5, and set cal.iniFixedP = ./readscf.h5.
When continuing a calculation, the number of electrons and the target electrode potential are obtained from the specified file; modifying these parameters in the in file will have no effect.
7.5 Read rho and wave restart instructions
Due to the computational expense of hybrid functional calculations, when a calculation fails to converge in one step or when higher convergence accuracy is desired, the already obtained charge density and wave function files can be read. This is achieved by specifying the file paths using the cal.iniCharge and cal.iniWave parameters. The following Resatrt-HSE.in file lists the key parameters for restarting a hybrid functional calculation:
# task type
task = scf
#hybrid related
sys.hybrid=true
sys.hybridType=HSE06
#read related
cal.iniCharge = ../01/rho.bin
cal.iniWave = ../01/wave.bin
#outputs related
io.charge = true
io.wave = true
Note:
For hybrid functional calculations, both the charge density and wave function files are required for continuation, and neither can be omitted.
For hybrid functional calculations, it is recommended to output the rho.bin and wave.bin files, which can be used as input for continuation calculations.